Principe de précaution sur la thérapie génique

Suite à la survenue d’évènements indésirables graves et inattendus, bluebird bio a suspendu le mois dernier deux essais cliniques et la commercialisation de son premier médicament, Zynteglo®. Le point sur la situation et les dernières données, « rassurantes », avec le Dr Fabrizia Bignami, directrice médicale France de bluebird bio.
Une survenue de leucémie aiguë myéloïde chez un patient traité plus de cinq ans auparavant par sa thérapie génique expérimentale Lentiglobin, une suspicion de syndrome myélodysplasique chez un autre patient traité en 2020 lors du même essai… Mi-février, bluebird bio décide non seulement de suspendre son programme clinique dans la drépanocytose mais aussi la commercialisation de sa première thérapie génique, autorisée en Europe en mai 2019, Zynteglo® (dans la bêta-thalassémie transfusion-dépendante), utilisant le même vecteur. Même si les premiers résultats d’analyse divulgués dès le 10 mars par le laboratoire tendent à dédouaner la thérapie génique, ce cas illustre la nécessité de rester vigilant quant aux effets à long terme de ces technologies.
Pourquoi la survenue d’un cancer chez un patient a-t-elle conduit à soupçonner une thérapie génique administrée des années auparavant ?
Dr Fabrizia Bignami, bluebird bio : Notre approche est celle d’une thérapie génique intégrative ex-vivo. Les cellules souches du patient sont prélevées ; notre vecteur lentiviral, BB305, permet l’insertion du gène thérapeutique dans le génome de ces cellules, où il doit demeurer à vie. Réinjectées, les cellules souches se différencient ensuite en cellules sanguines portant un gène fonctionnel. Le risque évoqué ici est celui que l’insertion du vecteur puisse « casser » quelques gènes et en altérer le fonctionnement, provoquant ce que l’on appelle une oncogenèse insertionnelle. Ce type d’événement indésirable grave est déjà survenu lors des tout débuts de la thérapie génique. Depuis cette époque, grâce au travail combiné des chercheurs, des cliniciens, mais aussi des autorités réglementaires, nous en sommes aujourd’hui à la troisième génération de vecteurs lentiviraux, structurés de façon à ne pas déclencher ce genre d’effet indésirable. Mais en sciences, nous ne sommes jamais à l’abri d’une surprise… Aussi tous nos protocoles cliniques prévoient, si un événement indésirable de ce type est observé, de suspendre les études et d’examiner au niveau moléculaire si le vecteur a pu ou non en être l’élément déclencheur. Et c’est exactement ce que nous avons fait, en mettant à l’arrêt notre programme clinique dans la drépanocytose. Par principe de précaution, nous avons aussi décidé de mettre en suspens la commercialisation de Zynteglo®, notre traitement de la bêta-thalassémie, même si dans cette indication, avec jusqu’à sept ans de recul, nous n’avons encore jamais observé ce type d’événement chez les 63 patients traités lors des essais cliniques.
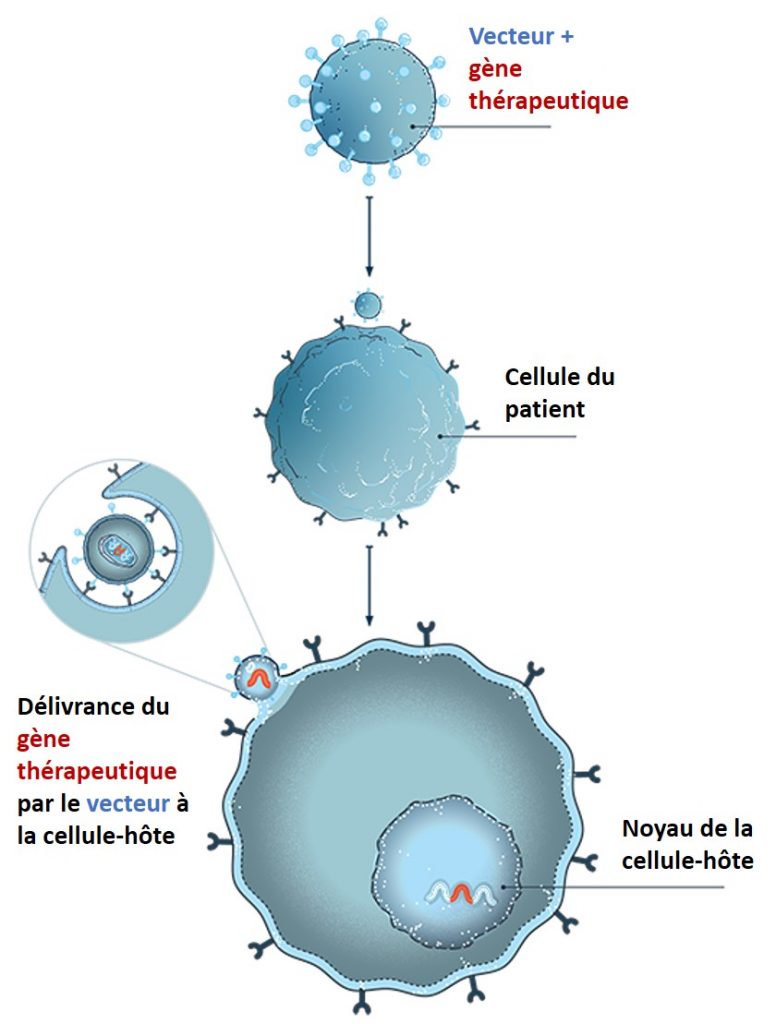
N’est-ce pas pousser un peu loin ce principe de précaution ?
Notre priorité est la sécurité des patients. Ces opérations de contrôle ne sont pas du tout inhabituelles dans le champ de la thérapie génique, pour écarter toute implication éventuelle des vecteurs : même si elle démontre une efficacité majeure, cela reste une technologie nouvelle. Être aussi transparent sur les risques potentiels que sur les bénéfices, c’est rendre service à toute la filière des thérapies géniques. Si nous n’avions pas pris ces précautions, on nous l’aurait reproché : il suffit de voir les réactions concernant les vaccins anti-Covid !

Dr Fabrizia Bignami, directrice médicale France de bluebird bio :
« Être aussi transparent sur les risques potentiels que sur les bénéfices, c’est rendre service à toute la filière des thérapies géniques. »
Comment comptez-vous lever les doutes ?
Nous avons très rapidement réalisé les analyses moléculaires en laboratoire. Nous avons déjà pu partager la semaine dernière des résultats que nous jugeons très concluants. Concernant le cas de leucémie aiguë myéloïde, nous avons notamment recherché quel était le site d’insertion du vecteur dans le génome des cellules cancéreuses. Or le gène dans lequel il est inséré n’est pas connu pour être impliqué dans quelque type de cancer. Nous n’avons pas constaté non plus d’anomalie dans l’expression des gènes voisins du site d’insertion. Sur cette base, nous avons d’ores et déjà démarré les discussions avec les autorités réglementaires pour pouvoir relancer rapidement les essais cliniques, éventuellement après des analyses complémentaires si besoin.
Et concernant Zynteglo® ?
Suite à notre décision de suspendre la commercialisation, la Commission européenne a demandé à l’EMA de confirmer le rapport bénéfice-risque de Zynteglo® avant de reprendre le processus de renouvellement de notre AMM conditionnelle. A ce jour, un seul patient a été traité dans le cadre de la commercialisation du produit, en Allemagne. Une première réunion du PRAC (comité d’évaluation des risques de pharmacovigilance) a déjà eu lieu la semaine dernière. Une démarche de questions-réponses va être enclenchée. C’est un déroulement tout à faire classique dans ce type de cas, même s’il est difficile de donner une date de fin à ce stade. Mais nous sommes plutôt sereins.
Propos recueillis par Julie Wierzbicki







